The cell cytoskeleton plays a key role in human biology and disease, contributing ubiquitously to such important processes as embryonic development, wound repair and cancer metastasis. We are interested in gaining deeper understanding of the physical chemistry behind these complex, far-from-equilibrium mechanochemical processes. The approach and model, named Mechanochemical Dynamics of Active Networks (MEDYAN), is based on combining stochastic reaction-diffusion treatment of cellular biochemical processes with polymer physics of cytoskeletal filament network growth, while explicitly coupling chemistry and mechanics.
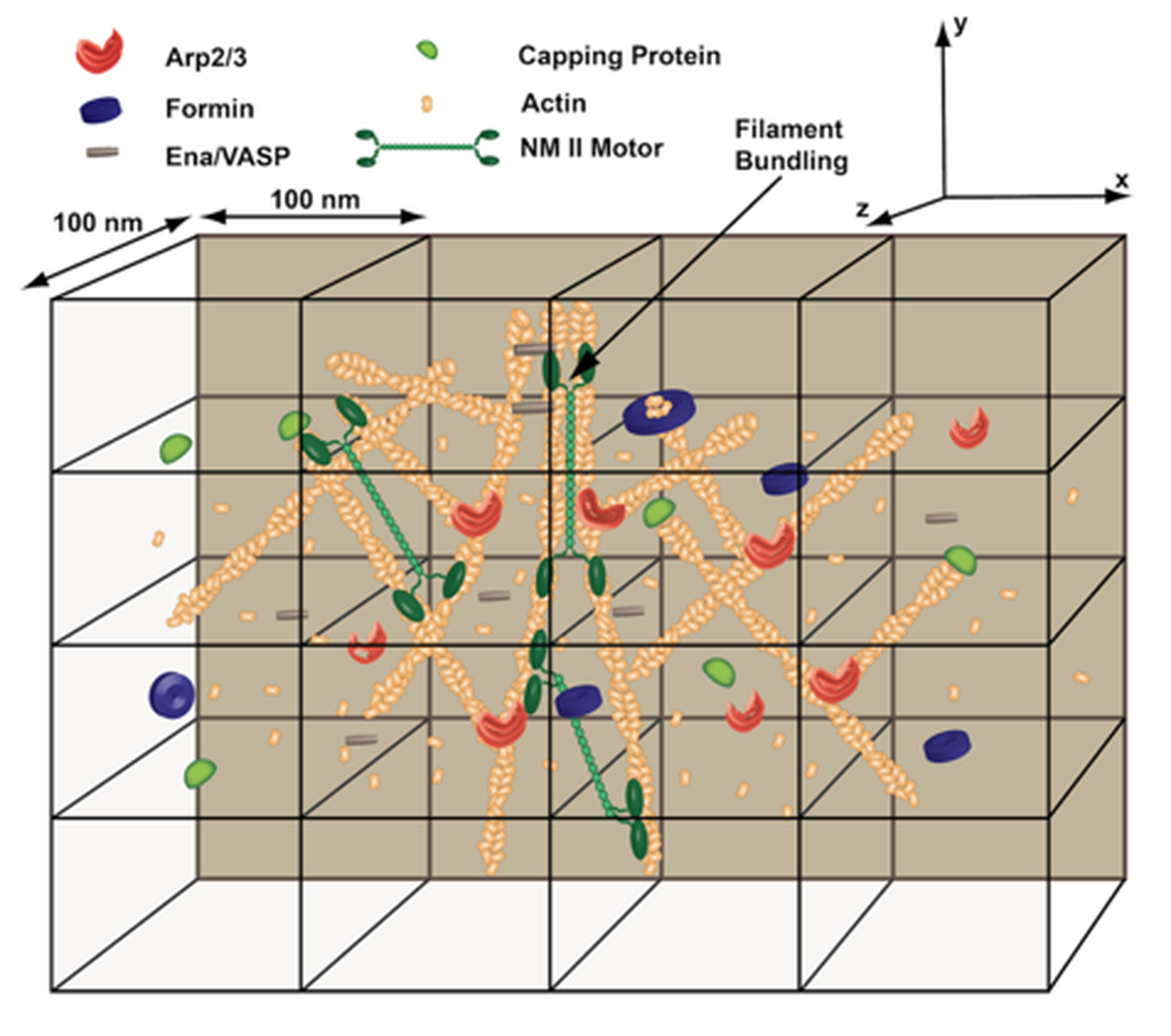
While most cytoskeletal models in literature emphasize either reaction-diffusion modeling of cytoskeletal chemistry or semi-flexible polymer chain mechanics of the constituent filaments, the MEDYAN model intimately couples the chemical processes of an active cytoskeletal network with its underlying polymer mechanics by iteratively swtiching between stochastic reaction-diffusion simulation and network mechanical equilibration, producing an efficient yet detailed cytoskeletal network evolution. With chemical and mechanical representations of semi-flexible polymers, cross-linking molecules, molecular motors, as well as polymer branching and severing molecules, MEDYAN allows for an explicit treatment of the key elements involved in cytoskeletal evolution.
Our lab has developed a third-generation C++ software package based on the MEDYAN model, to simulate growth dynamics of actin based filamentous networks in vitro and in vivo. This code is publicly available for scientific use at
medyan.org, which contains the MEDYAN source code, installation instructions, visualization tools, and user guide.
Proteins and other biomolecules can be simulated on a computer primarily in two ways: relying either on atomistic or coarse-grained force fields. The latter approaches, which enable treating large systems at long timescales, come in with rather broad variety of choices, compared with the atomistic models. In particular, many useful coarse-grained protein force fields require prior knowledge of the native structure for the specific protein target, often with the stated goal of simulating protein folding kinetics and dynamics.
These methods, however, cannot be used when the target protein structure is unknown or non-native interactions play a significant role. To address such problems, several coarse-grained protein force fields have been developed in the last 30 years or so that allow de novo structure prediction even in the absence of sequence homology.
The AWSEM potential is a prominent member of this latter group, where various research groups have successfully demonstrated its broad applications in monomeric protein structure prediction, binding predictions of dimers and multimeric assemblies, folding of membrane proteins, and structural and kinetic studies of protein-DNA complexes. AWSEM naturally covers the whole spectrum between native-structure-based and de novo methods, and can be used both with the knowledge of native structures, and without such explicit knowledge. In the latter case it relies solely on sequence information. Because AWSEM's potential is analytical and differentiable, it is currently implemented as a molecular dynamics algorithm (AWSEM-MD).
AWSEM is immplemented in C++ as an add-on package compatible with the LAMMPS simulation platform. This code is publicly available for scientific use at
awsem-md.org, which contains the AWSEM source code and user guides.